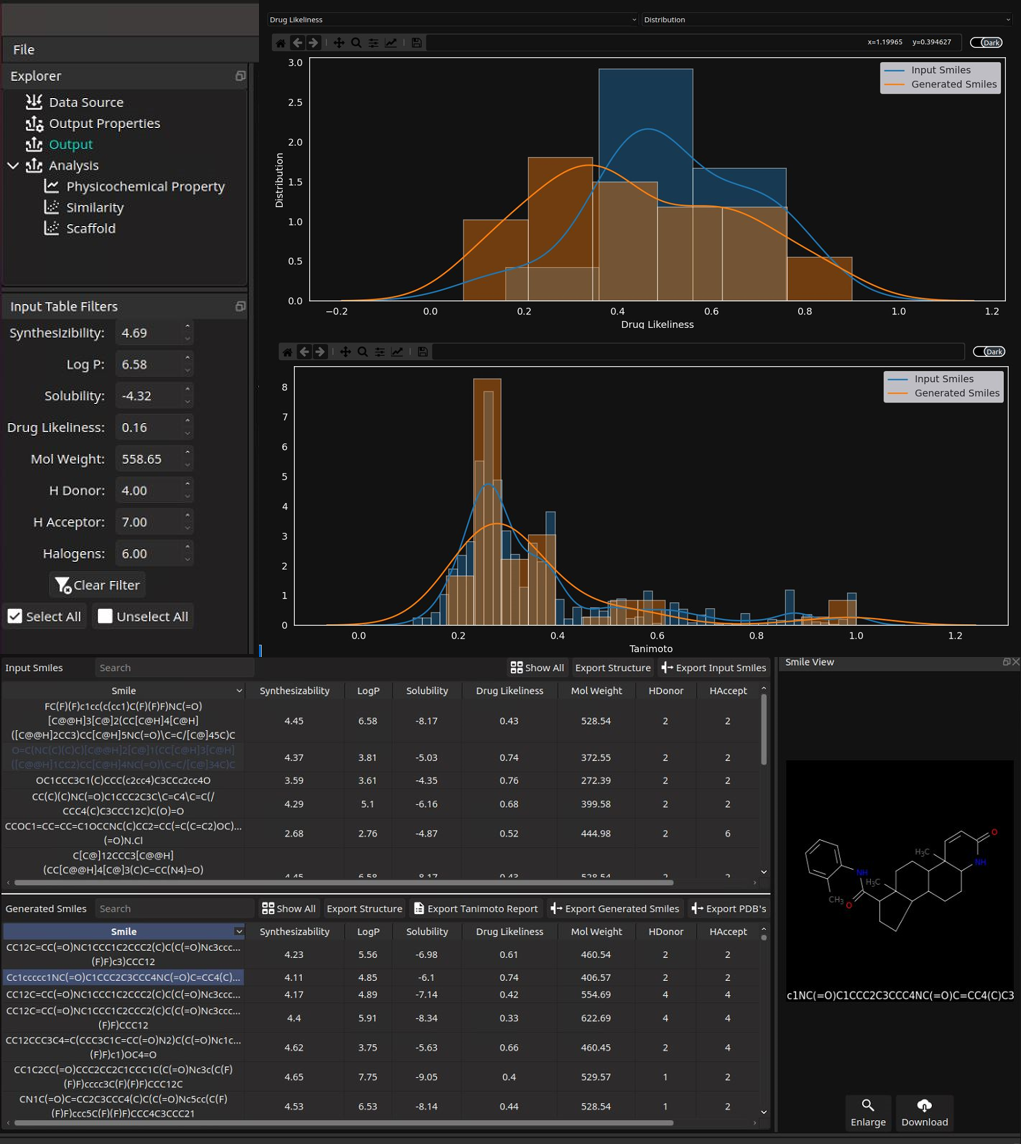
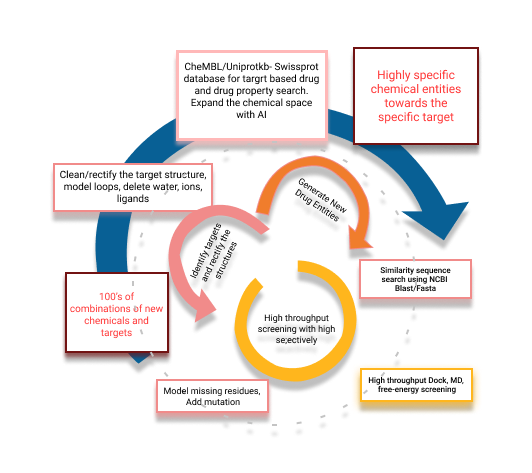
- Discover lead-like compounds from gigantic chemical space
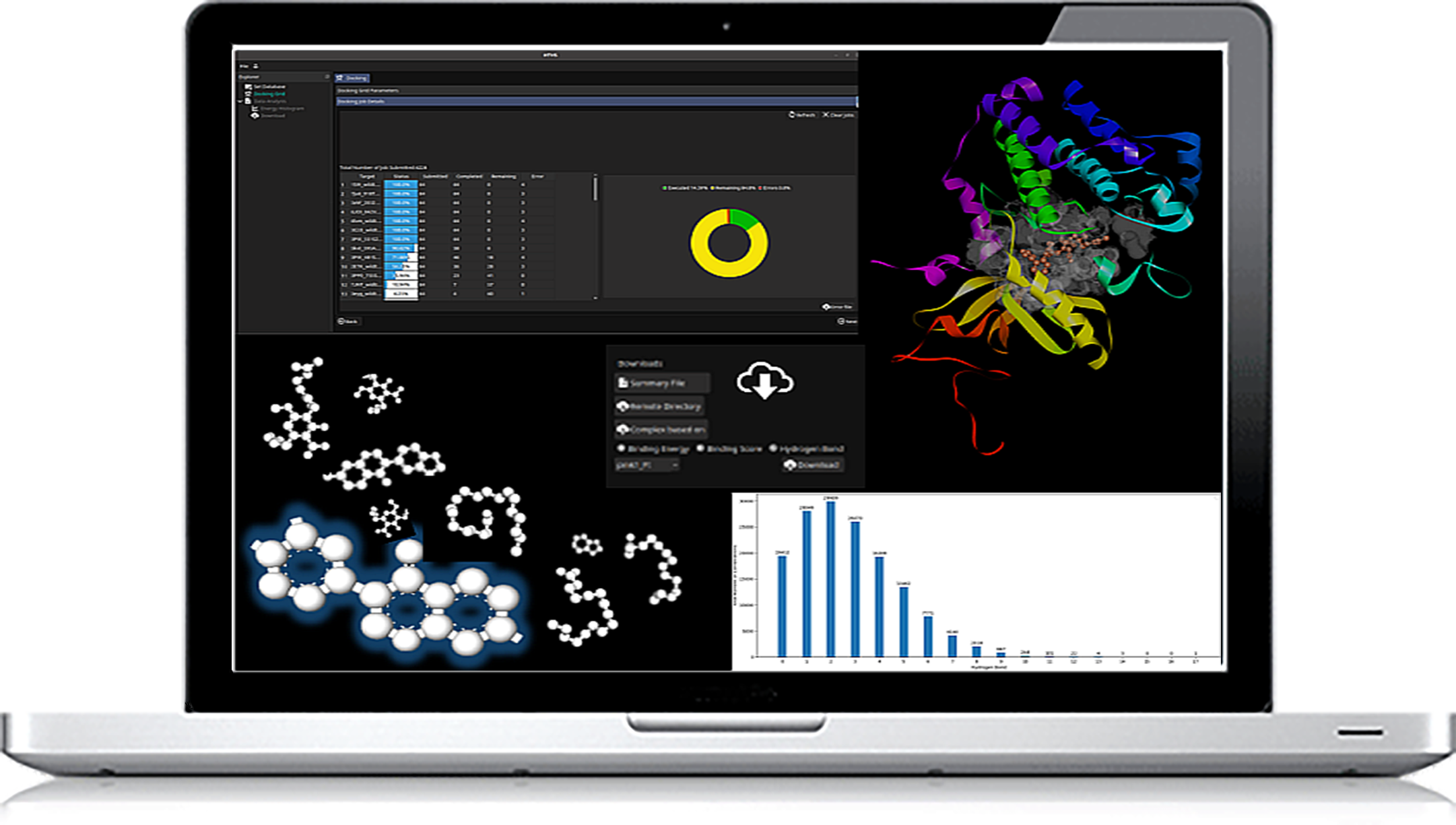
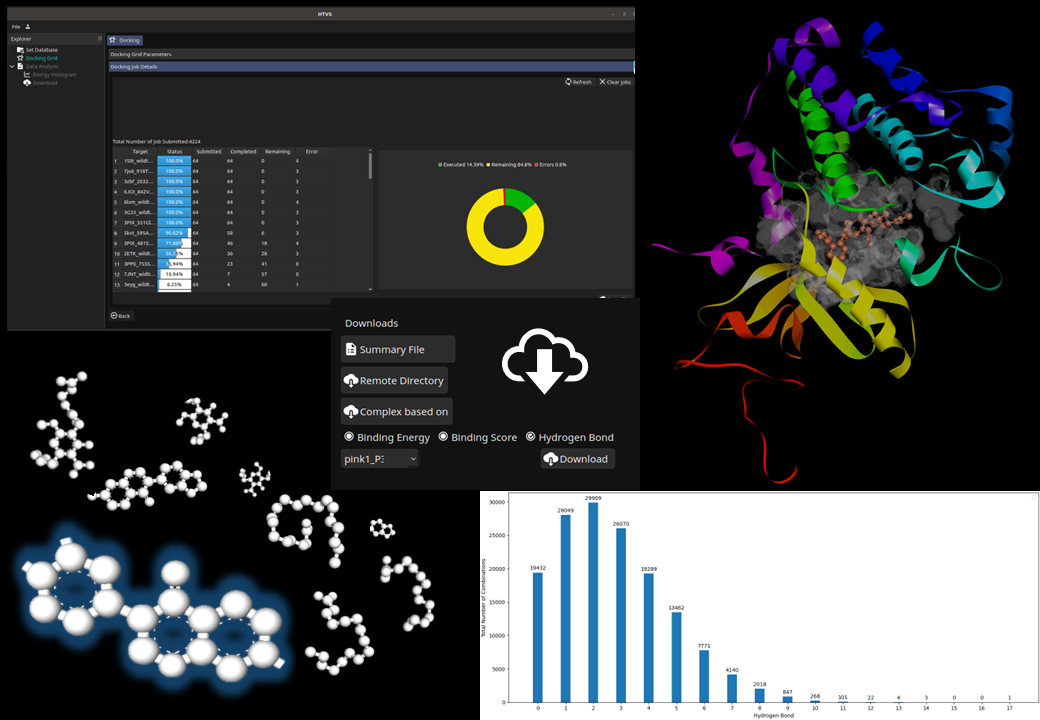
- Docking-based app to screen multiple targets with Thousands to Millions of compounds at one go
- Run calculations on cloud and/or in-house servers
- Options for algorithms - Lamarckian Genetic Annealing (LGA) and Local Search (LS)
- Choices of partial charges, Model type (Bound, Extended), Number of GA runs (Coarse grain, Highly coarse grain, fine-grain and user-defined) etc.
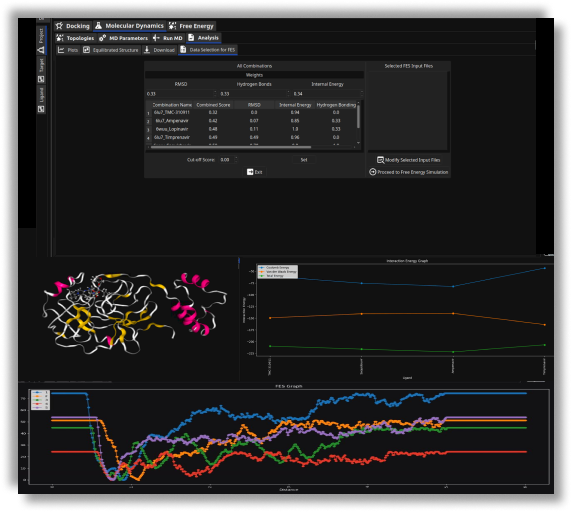
- Control output with docking scores, binding energy and H-bonds represented by histogram plots
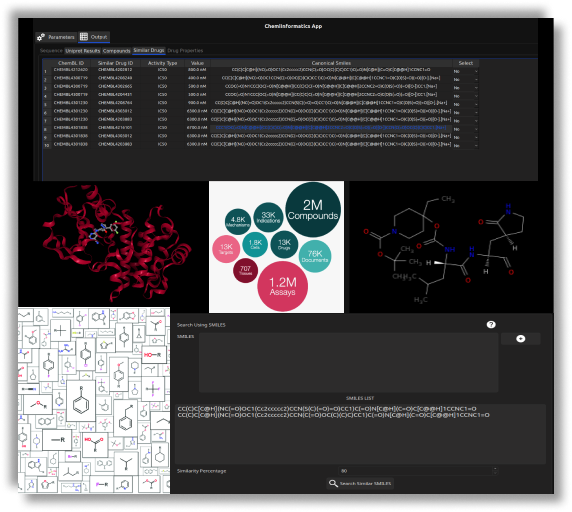
- Investigate the binding mode of lead-like compounds by further downstream refinement with the X-ESS app.